





 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste O Seminário Retina Bahia, com o tema “Visibilidade e Inclusão”, teve início na Câmara de Vereadores de Brumado nesta sexta-feira (15). Representando o Fórum Baiano de Pessoas Cegas e Com Baixa Visão, a Associação Baiana Para Arte e Cultura Inclusiva e o Movimento Brasileiro de Mulheres Cegas e Com Baixa Visão, Rita Ribeiro foi uma das palestrantes do seminário. Ao site Achei Sudoeste, ela defendeu que todos os segmentos devem estar presentes nos espaços para que o governo garanta as políticas públicas, a assistência necessária e os direitos de cada público, nesse caso as pessoas com deficiência visual. “A gente tem tido muitas perdas e nós precisamos ser incluídos na sociedade de fato. Me considero uma pessoa normal, a minha deficiência física é apenas uma condição, que não me impede de ir e vir, de ser ou não ser”, destacou. Segundo Ribeiro, o Retina Bahia, um grupo virtual formado por pessoas com degeneração na retina que procura apoiar outras pessoas com essa deficiência, busca a garantia de direitos e a inclusão desse público em específico. “A sociedade civil precisa nos entender e nos incluir de fato”, reiterou.
 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste A Agência Nacional de Vigilância Sanitária (Anvisa) autorizou na sexta-feira, 1º, o início de uma nova etapa do ensaio clínico com a SpiN-Tec - a primeira vacina 100% nacional contra a covid-19, desenvolvida pelo Centro de Tecnologia de Vacinas (CTVacinas) da Universidade Federal de Minas Gerais (UFMG). O objetivo é que o imunizante esteja disponível até o fim de 2024. Em nota, a Anvisa informou que o início da primeira fase do ensaio clínico foi autorizado pela agência em outubro do ano passado e, com base nos resultados preliminares de segurança e imunogenicidade obtidos, foi autorizado o prosseguimento do teste. “O objetivo dessa nova fase é obter dados adicionais de segurança e imunogenicidade, utilizando a dose que apresentou o melhor desempenho na primeira fase”, informa a nota. Nessa nova fase serão incluídas 372 pessoas saudáveis de ambos os sexos, com idade entre 18 e 85 anos, que já tenham completado o esquema vacinal primário contra a covid-19 e pelo menos uma dose de reforço há pelo menos seis meses. Pessoas que tiveram covid-19 há pelo menos seis meses também podem participar do estudo. O ensaio clínico, de acordo com a Anvisa, está sendo realizado em três centros em Belo Horizonte: na Faculdade de Medicina da UFMG, no Centro Freire de Pesquisa Clínica e no Centro Infection Control. A segunda etapa vai começar tão logo os voluntários sejam reunidos pelos centros de pesquisa. Os resultados da primeira fase de testes da SpiN-Tec foram divulgados pela Fiocruz em maio deste ano. Os dados preliminares não indicaram problemas de segurança e apontaram resposta imunológica ao vírus. A vacina SpiN-Tec consiste na fusão de duas proteínas (S e N), que resultam em uma proteína quimera. Essa associação confere à nova vacina um diferencial em relação aos demais imunizantes, que contemplam apenas a proteína S. Desta forma, os desenvolvedores do novo imunizante esperam oferecer uma proteção mais ampla contra possíveis novas variantes.
A miopia infantil é um distúrbio visual que afeta crianças, resultando em dificuldade para enxergar objetos distantes com clareza. De acordo com o Centro de Visão da Bahia (Cevib), os sintomas da miopia infantil podem incluir dificuldade em enxergar objetos distantes, como quadros na escola ou placas de trânsito, aproximar-se demais da TV, livros ou telas de dispositivos eletrônicos para conseguir ver melhor, apertar os olhos ou franzir a testa para tentar focar em objetos distantes e dor de cabeça frequente, especialmente após esforço visual prolongado. Se houver qualquer suspeita de que a criança possa estar desenvolvendo miopia é importante agendar uma consulta. Em Brumado, o Centro de Visão da Bahia fica localizado na Rua Euclides da Cunha, 175, Centro. Os telefones são: (77) 3441-6967 e (77) 99929-6967.
 Foto: Rodrigo Moraes
Foto: Rodrigo Moraes Faustão encontra-se sob cuidados intensivos e, em virtude do agravamento do quadro, há indicação para transplante cardíaco. As informações são do jornal o Globo. É o que diz o último boletim divulgado pelo Hospital Albert Einstein, às 19h24 deste domingo (20). Faustão está internado no hospital de São Paulo desde 5 de agosto, com quadro de insuficiência cardíaca. Em junho deste ano, deixou o comando de seu programa na Band, após um ano e meio. A nota, assinada pelos médicos Fernando Bacal e Miguel Cendoroglo Neto, traz as seguintes informações: “Em 05 de agosto, Fausto Silva deu entrada no Hospital Israelita Albert Einstein para tratamento de insuficiência cardíaca, condição que vem sendo acompanhada desde 2020. Ele encontra-se sob cuidados intensivos e, em virtude do agravamento do quadro, há indicação para transplante cardíaco. O paciente está em diálise e necessitando de medicamentos para ajudar na força de bombeamento do coração. Fausto Silva já foi incluído na fila única de transplantes, regida pela Secretaria de Estado da Saúde de São Paulo, que leva em consideração, para definição da priorização, o tempo de espera, a tipagem sanguínea e a gravidade do caso”.
 Foto: Reprodução/TV Band
Foto: Reprodução/TV Band O apresentador Fausto Silva está internado no Hospital Albert Eistein, em São Paulo, há quase duas semanas. Em nota, o centro médico informou que o apresentador está no local desde o último dia 5 de agosto fazendo um “tratamento de compensação clínica de insuficiência cardíaca”. Neste momento, encontra-se estável e em cuidados intensivos”, diz a nota. Segundo o portal Metrópoles, o quadro do apresentador não é grave. Luciana Cardoso, esposa de Faustão, confirmou a internação, mas não detalhou o motivo que levou o ex-Globo ao hospital.
 Foto: Divulgação/PRF
Foto: Divulgação/PRF Na segunda-feira (07), a Polícia Rodoviária Federal (PRF) realizava fiscalização de trânsito e combate à criminalidade no km 830 da BR-116, no município de Vitória da Conquista, na região sudoeste da Bahia, quando abordou um ônibus de viagem que seguia de São Paulo com destino a Natal. Durante vistoria no bagageiro externo do veículo, foram encontrados 24 frascos de um certo tipo de produto, os quais não apresentavam a sua composição e procedência nos rótulos. Apenas alegavam indicações para tratamento das mais diversas patologias, como artrite, artrose, doenças da pele, gastrite, colesterol e osteoporose. As mercadorias foram despachadas por encomenda e não possuíam registro na Agência Nacional de Vigilância Sanitária (Anvisa). A princípio, a PRF constatou a ocorrência de falsificação, corrupção, adulteração ou alteração de produto destinado a fins terapêuticos ou medicinais. Os medicamentos foram encaminhados para a Polícia Civil de Vitória da Conquista para abertura do inquérito policial e continuidade das investigações. O ônibus foi liberado para seguir viagem.
 Foto: Freepik
Foto: Freepik A ejaculação precoce é uma das disfunções sexuais mais prevalentes: ela acomete 1 a cada 3 brasileiros acima de 18 anos, segundo dados da Sociedade Brasileira de Urologia (SBU). As informações são do jornal o Globo. Recentemente, a Agência Nacional de Vigilância Sanitária (Anvisa) aprovou o primeiro medicamento específico para o problema com indicação em bula e uso sob demanda. Essa medicação já é usada em mais de 50 países, como Itália, Reino Unido, Austrália, Argentina e Uruguai. Pessoas com ejaculação precoce normalmente chegam ao orgasmo em menos de um minuto. O tempo ejaculatório considerado normal, do momento da penetração até a ejaculação, é de três a seis minutos. A disfunção está, em muitos casos, relacionada à ansiedade. Há dois tipos de ejaculação precoce, a primária, quando o paciente sofre com isso desde o primeiro relacionamento, e a secundária, quando ele passa a apresentar uma ejaculação mais rápida como efeito colateral a algum fator ocorrido, de fundo psicológico ou físico. A dificuldade de manter uma relação sexual por mais tempo acaba trazendo prejuízos emocionais para o paciente, assim como problemas para o relacionamento. Com o princípio ativo de cloridrato de dapoxetina (com o nome comercial de Prosoy, comercializado pela Farmoquímica), o remédio deve ser tomado de uma a três horas antes da relação sexual, por isso é considerado sob demanda. Antes da chegada do novo medicamento, as únicas alternativas de tratamento para a ejaculação precoce eram com o uso ansiolíticos e antidepressivos, por conta do fundo emocional do problema. Os fármacos eram de uso contínuo e demoravam quase 1 mês para começar a surtir algum efeito. O remédio deve ser prescrito por um médico e sua venda é controlada (tarja vermelha). Ele é vendido em comprimidos em dois tipos de dosagens: de 30 e 60mg. No entanto, a bula recomenda que o paciente comece com a dose de 30mg tome, no máximo, um comprimido a cada 24 horas.
 Foto: Fabio Rodrigues Pozzebom/Agência Brasil
Foto: Fabio Rodrigues Pozzebom/Agência Brasil A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou nesta segunda-feira (26) o registro da vacina bivalente da Moderna contra a Covid-19. É o primeiro registro definitivo para um imunizante bivalente no Brasil. As informações são do Bahia Notícias, parceiro do Achei Sudoeste. A vacina está indicada para crianças a partir de 6 anos e adultos como dose de reforço -ou seja, só pode ser aplicada em quem já se vacinou contra a doença. Segundo a Anvisa, a Spikevax é uma vacina bivalente que tem proteção contra a cepa original Wuhan e contra a cepa Ômicron do tipo BA4 e BA5. O pedido de registro da vacina bivalente da Moderna foi encaminhado à Anvisa em janeiro de 2023 pela Adium, empresa responsável pela comercialização da vacina no país. Para analisar a vacina, a Anvisa contou com o apoio de especialistas externos, como a SBI (Sociedade Brasileira de Imunologia), a SBP (Sociedade Brasileira de Pediatria), a SBI (Sociedade Brasileira de Infectologia), a SBIm (Sociedade Brasileira de Imunizações) e a SBPT (Sociedade Brasileira de Pneumologia e Tisiologia). De acordo com a Adium Brasil, o próximo passo é a definição do preço pela CMED (Câmara de Regulação do Mercado de Medicamentos) e a posterior análise pela Conitec (Comissão Nacional de Incorporação de Novas Tecnologias no SUS), que determina se recomenda a incorporação da vacina ao sistema público de saúde. “Estamos muito satisfeitos em (...) poder em breve proporcionar à população brasileira o acesso a uma imunização eficaz com tecnologia de mRNA em sua concepção”, destaca Alexandre Seraphim, CEO da Adium no Brasil. A Spikevax bivalente já está autorizada em 38 países. O uso da vacina foi aprovado pela EMA, agência europeia de medicamentos, e pela FDA, agência reguladora dos Estados Unidos (Food and Drug Administration - FDA), entre outras.
 Foto: Reprodução/G1
Foto: Reprodução/G1 Uma nova vacina contra a dengue estará disponível no Brasil a partir da semana que vem, segundo a Associação Brasileira de Clínicas de Vacinas (ABCVAC). A Qdenga (TAK-003), do laboratório japonês Takeda Pharma, é o primeiro imunizante liberado no país para pessoas que nunca entraram em contato com o vírus da dengue. Neste primeiro momento, a Qdenga será aplicada apenas na rede privada, como clínicas, laboratórios e farmácias. O valor de cada dose deve ficar entre R$ 301,27 e R$ 402,05, segundo preço tabelado pela Câmara de Regulação do Mercado de Medicamentos (CMED). “As clínicas devem utilizar esse parâmetro na composição da sua precificação final, que também inclui o atendimento, a triagem, a análise da caderneta de vacinação, as orientações pré e pós-vacina, além de todo o suporte que os pacientes necessitam para se informar corretamente sobre a questão da vacinação”, disse a ABCVAC, em nota enviada ao G1. O registro da Qdenga foi aprovado pela Agência Nacional de Vigilância Sanitária (Anvisa) em março deste ano.
 Foto: Divulgação
Foto: Divulgação  Foto: Geovana Albuquerque/Agência Saúde DF
Foto: Geovana Albuquerque/Agência Saúde DF A Agência Nacional de Vigilância Sanitária (Anvisa) divulgou uma nota nesta sexta-feira (17) na qual atesta que as vacinas bivalentes BA.1 e BA.4/BA.5 contra a Covid-19, produzidas pela empresa Pfizer, estão dentro do prazo de validade e, portanto, podem ser utilizadas com segurança. No documento, a Anvisa destaca que os imunizantes podem ser utilizados dentro do prazo de 18 meses, a partir da data de fabricação dos produtos. “Anteriormente aprovadas para uso em até 12 meses, essas vacinas passaram por um rigoroso processo de avaliação técnica da agência de estudos de estabilidade, antes da aprovação da ampliação do prazo de validade”, diz a nota. A avaliação dos dados dos estudos demonstrou ainda, segundo a Anvisa, não haver alteração nas especificações de qualidade das vacinas no período adicional ao prazo anteriormente autorizado. “As vacinas são seguras, eficazes e podem ser utilizadas pelo Programa Nacional de Imunizações do Ministério da Saúde, conforme os estudos de estabilidade avaliados e aprovados pela Agência”, garante a diretora Meiruze Sousa Freitas. Sobre a ampliação do prazo de validade, a Anvisa ressalta que ela é permitida mediante medidas de comunicação e de rastreabilidade dos lotes, adotadas pela Pfizer. Entre essas medidas está a inclusão, no portal eletrônico da Pfizer e no portal eletrônico Comirnaty Education, da listagem de todos os lotes disponíveis no Brasil e dos seus respectivos prazos de validade, para consulta dos cidadãos e profissionais de saúde envolvidos na aplicação das vacinas. Os cuidados de conservação não sofreram alterações.
 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste A Qdenga, nova vacina utilizada para prevenção da dengue, deve chegar ao Brasil somente no segundo semestre deste ano. Fabricada pela Takeda, ela foi aprovada no início deste mês pela Agência Nacional de Vigilância Sanitária (Anvisa) para ser comercializado no Brasil. O imunizante tem quatro diferentes sorotipos do vírus causador da doença, conferindo assim uma ampla proteção. Destinado ao público de 4 a 60 anos, ele é aplicado em esquema de duas doses, com intervalo de três meses entre elas. Segundo Vivian Lee, diretora-executiva de assuntos médicos da Takeda Brasil, a previsão estimada para o produto estar em unidades de saúde do país leva em consideração trâmites necessários para a sua comercialização. Depois da aprovação da Anvisa, outra etapa se dá na CMED (Câmara de Regulação do Mercado de Medicamentos), que define o valor máximo de medicamentos. Por enquanto, não há estimativa de quanto a dose deve custar. Lee diz que a vacina já foi submetida à câmara e que agora é necessário aguardar. Segundo a diretora, essa etapa normalmente dura em torno de três meses, porém existe a possibilidade de levar mais tempo. Na perspectiva mais otimista, de o processo durar três meses, Lee observa que a vacina deve estar disponível no Brasil no segundo semestre deste ano. Segundo Lee, a farmacêutica tem capacidade de distribuir a vacina no setor privado no país, mas os pedidos ainda não foram feitos pelas clínicas por ainda não haver o valor definido do fármaco. “Não temos recebido negociação porque não temos a definição do preço na CMED”, afirma a diretora. Também existe o interesse de levar o imunizante para o SUS, até por causa do impacto da dengue no Brasil. Só no ano passado houve mais de mil mortes.
 Foto: Lay Amorim/Achei Sudoeste
Foto: Lay Amorim/Achei Sudoeste A diretoria da Agência Nacional de Vigilância Sanitária (Anvisa) decidiu retirar a obrigatoriedade de uso de máscara dentro de aviões e aeroportos no Brasil. A medida foi publicada nesta quarta-feira (1º). Em novembro do ano passado, a Anvisa havia decidido sobre a volta da obrigatoriedade das máscaras. De acordo com as informações, a obrigatoriedade só deve continuar para o tripulante que esteja com caso suspeito de doença. Apesar disso, o desembarque por fileira foi mantido. Para a Anvisa, a medida é um legado da pandemia e que também ajuda a evitar tumulto na saída das aeronaves.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil Um pedido de Autorização de Uso Emergencial para a versão da vacina bivalente contra a Covid-19, desenvolvida pelo laboratório farmacêutico Moderna e comercializada pela Adium, foi apresentado à Agência Nacional de Vigilância Sanitária (Anvisa), nesta sexta-feira (17). Segundo a Anvisa, a vacina bivalente contém uma mistura de cepas do vírus SARS-Cov-2 e promete conferir maior proteção à variante Ômicron, quando comparada com vacinas monovalentes. Por sua transmissibilidade, observa a agência, a variante Ômicron causa preocupação às autoridades sanitárias do país. A Adium havia apresentado o pedido de registro da vacina, em janeiro. “Este pedido se encontra em análise pela equipe técnica da Anvisa. Porém, a empresa decidiu protocolar a Autorização de Uso Emergencial, paralelamente ao pedido do registro, de acordo com a manifestação favorável do Ministério da Saúde, conforme previsto no parágrafo único do Art. 1º da Resolução (RDC) 688, de 13 de maio de 2022”, explicou a agência, em nota.
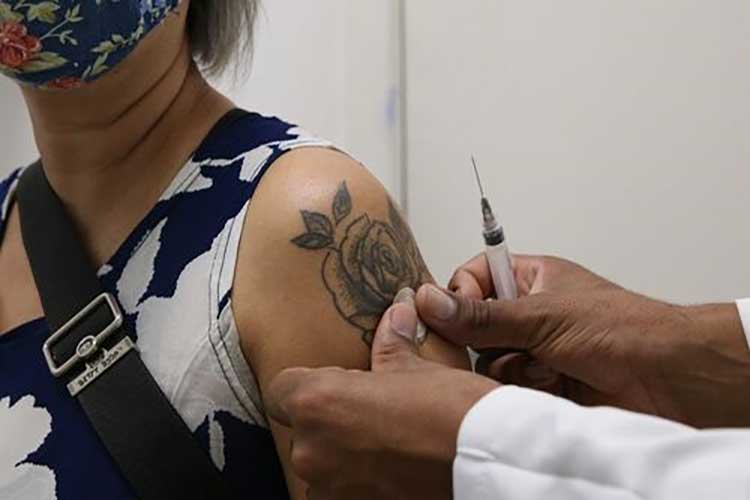 Foto: Rovena Rosa/Agência Brasil
Foto: Rovena Rosa/Agência Brasil O Ministério da Saúde vai iniciar campanha de reforço contra a Covid-19 com vacinas bivalentes a partir do dia 27 de fevereiro. A primeira fase de vacinação será direcionada para os grupos prioritários de pessoas acima de 70 anos, imunocomprometidos e comunidades indígenas, ribeirinhas e quilombolas. De acordo com o Brasil 61, 0 plano de imunização para 2023 foi divulgado nesta quinta-feira (26), na primeira reunião da Comissão Intergestores Tripartite (CIT) do ano. A ministra da Saúde, Nísia Trindade, defende um “movimento nacional” com participação da sociedade e dos governos federal e estaduais. Ela ressalta a importância de uma ação estratégica coordenada. “Estou muito confiante nessa ação de vacinação, mas sabendo que ela é muito complexa. Não deveríamos ter tantos problemas de confiança, mas temos. Então vamos trabalhar para reduzir todos os gargalos. A resposta não será única. O Brasil é muito diverso. Alguns municípios avançaram muito na vacinação e muito bem, outros não. Olhar esse diagnóstico”, afirma a ministra. As vacinas bivalentes são aquelas que oferecem proteção contra mais de uma cepa de determinado vírus. Em novembro de 2022, a Agência Nacional de Vigilância Sanitária (Anvisa) aprovou o uso temporário e emergencial de dois imunizantes da empresa Pfizer contra a Covid-19.
 Foto: Reprodução/Pfizer
Foto: Reprodução/Pfizer A aprovação, pela Agência Nacional de Vigilância Sanitária (Anvisa), de um novo medicamento para tratamento da obesidade, trouxe uma esperança para pacientes que buscam tratamento para perda de peso. A semaglutida já era utilizada no Brasil para tratamento do diabetes tipo 2 e agora pode ser usada também no tratamento de sobrepeso e obesidade, em forma de medicamento injetável. Há, no entanto, diferença de dosagem. Enquanto que para o controle da glicose a semaglutida aplicada é de 0,5 miligrama a 1 miligrama, para redução do peso corporal a substância é de 2,4 miligramas. A semaglutida é da mesma família da liraglutida, também utilizada nos dois tratamentos. Mas o diferencial do medicamento aprovado pela Anvisa no início deste ano é sua eficácia. A semaglutida é vista entre os médicos como um avanço no tratamento da obesidade. Isso porque os outros medicamentos existentes possibilitam uma perda de peso de, no máximo, 10%. “Quando o paciente tem uma indicação de perda de peso inferior a 5% do peso corporal, a melhor indicação é mudança de hábito alimentar. Quando tem a necessidade de perda de peso de mais de 5%, até 15%, aliam-se as mudanças de hábitos de vida e a terapia farmacológica. A novidade é que a semaglutida pode reduzir mais de 15%”, explica o médico Paulo Miranda, presidente da Sociedade Brasileira de Endocrinologia e Metabologia (SBEM).
 Foto: JC Gellidon/Unsplash
Foto: JC Gellidon/Unsplash A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou nesta sexta-feira (16) por unanimidade o uso emergencial do medicamento Evusheld (ou AZD7442), da AstraZeneca, para tratamento da Covid-19. O medicamento possui uma combinação de anticorpos monoclonais cilgavimabe + tixagevimabe e era indicado para indivíduos que não estão infectados com a Covid-19 e não tiveram contato com o vírus. De acordo com a agência reguladora, o medicamento é indicado para pacientes com 12 anos ou mais com Covid-19 que não necessitam de oxigênio suplementar e que demonstrem risco aumentado de progressão para o estado grave da doença. O medicamento também é indicado para pessoas que não devem tomar a vacina da Covid-19 devido a um histórico de reação adversa grave. Para quem pode usar o imunizante, ele deve ser administrado pelo menos duas semanas após a vacinação. O Evusheld já foi aprovado por outras agências reguladoras em países como os Estados Unidos, França, Israel, Itália, Barein, Egito e Emirados Árabes Unidos.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil Após duas horas de sessão, a Agência Nacional de Vigilância Sanitária (Anvisa) formou maioria na noite desta terça-feira (22) para aprovar o uso emergencial de duas vacinas bivalentes contra a Covid-19. Três dos cinco diretores aprovaram o uso dos imunizantes produzidos pela Pfizer para proteger contra as subvariantes da Ômicron do novo coronavírus. A reunião ainda está em andamento, mas a autorização pode ser considerada aprovada porque a maioria dos diretores votaram a favor. A Anvisa autorizou a aplicação como doses de reforço em pessoas a partir de 12 anos, três meses depois da última dose de reforço. Consideradas de segunda geração, as vacinas bivalentes protegem contra a variante original do novo coronavírus, da Província de Wuhan (China), e contra as últimas subvariantes da Ômicron. Esta última é mais transmissível, porém mais branda, com o vírus se concentrando na garganta e não atingindo os pulmões. A variante original é menos contagiosa, porém mais perigosa e mais mortal. Os imunizantes bivalentes terão frascos na cor cinza para facilitar a identificação. As vacinas da Pfizer usam a tecnologia do RNA mensageiro, em que uma parte da proteína spike, responsável pela fixação do vírus nas células, é injetada para estimular a produção de anticorpos.
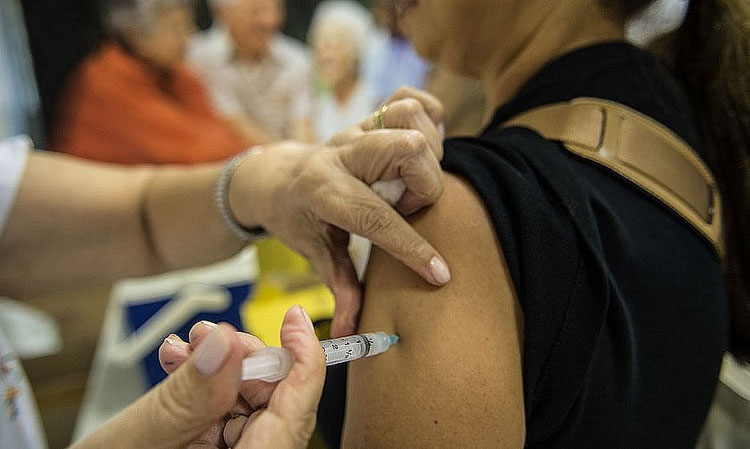 Foto: Agência Brasil
Foto: Agência Brasil A nova vacina que protege contra as mais recentes variantes de coronavírus ainda não tem data para ser liberada no Brasil. De acordo com nota divulgada pela Agência Nacional de Vigilância Sanitária (Anvisa), o imunizante passa por uma “fase final” de análises e deve ser liberado “em breve”, mas não há perspectiva de quando ele começará a ser aplicado no País. “Os processos estão em fase final de análise pela área técnica, para posterior envio à Diretoria da Agência para deliberação, considerando que se trata de autorização de uso emergencial. Não há ainda data fixada de decisão, mas a mesma deve ocorrer em breve”, declarou a autarquia. Segundo a Anvisa, “estão em fase final as análises das novas versões de vacina do laboratório Pfizer contendo as subvariantes BA.1 e BA.4 /BA.5?”. A agência diz ainda que a liberação depende de etapas como esclarecimentos dos fabricantes e discussão com sociedades médicas. Após uma diminuição de casos proporcionada pelo uso disseminado das vacinas, a ocorrência de infectados com coronavírus voltou a crescer no mundo todo. Na semana passada, a China registrou a maior quantidade de casos dos últimos seis meses. No Brasil, o número de confirmados com a doença também cresceu. No último dia 11, o País registrou 20.914, o maior número desde o dia 31 de agosto, quando foram registrados 61.085 com a doença. O aumento acontece depois do surgimento de novas subvariantes da variante Ômicron, que podem ser mais transmissíveis e resistentes às barreiras vacinais. Além da nova geração de vacinas, médicos recomendam o reforço com as terceiras e quartas doses das vacinas atuais Segundo médicos ouvidos pelo Estadão, na nova onda da Covid-19, a maioria dos hospitalizados é de pacientes idosos ou imunossuprimidos (pacientes transplantados, oncológico etc). Há também casos de pessoas com a vacinação atrasada - que não tomaram nenhuma dose ou deixaram de receber doses de reforço.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil A Agência Nacional de Vigilância Sanitária (Anvisa) aprovou o registro sanitário do terceiro produto de terapia gênica para tratamento de câncer. O Yescarta® (axicabtagene ciloleucel), fabricado pela Gilead?Sciences Farmacêutica do Brasil, é destinado ao tratamento de pacientes adultos com linfoma de grandes células B (LDGCB), recidivado ou refratário. A terapia com células geneticamente modificadas, segundo a Anvisa, tem demonstrado perfil de segurança e eficácia no tratamento de pacientes em recidiva e refratariedade para linfomas graves. “O produto aprovado é composto por células T autólogas com receptor de antígeno quimérico (CAR), projetadas para eliminar células tumorais que expressam CD19.”
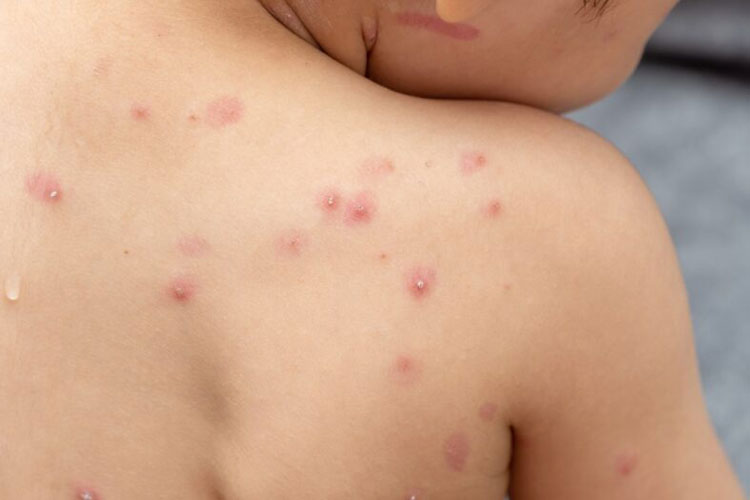 Foto: Divulgação
Foto: Divulgação A Agência Nacional de Vigilância Sanitária aprovou a importação e o uso do medicamento Tecovirimat, para o tratamento da varíola dos macacos, sem a necessidade de registro prévio. A decisão acata pedido feito pelo Ministério da Saúde. A dispensa foi dada em caráter excepcional e vigorará durante seis meses, podendo ser suspensa pela Anvisa. O remédio é o mesmo usado nos Estados Unidos e é produzido pela empresa americana Catalent Pharma Solutions. A autorização se refere ao Tecovirimat na concentração 200 mg, em cápsula. O medicamento é indicado para tratamento de doenças causadas por Orthopoxvírus, família da qual faz parte o monkeypox, em adultos, adolescentes e crianças com peso mínimo de treze quilos. A agência também aprovou a dispensa de registro para a importação e uso da vacina Jynneos / Imvanex, a única com ação específica contra a monkepox. O imunizante já está em uso nos Estados Unidos e na Europa e é indicado para adultos. A dispensa também é temporária, valendo por seis meses.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil O uso de máscaras de proteção facial deixa de ser obrigatório em aeroportos e aeronaves no Brasil. Diante do atual cenário, o uso de máscaras, adotado até então como medida de saúde coletiva, é convertido em medida de proteção individual. A determinação da Agência Nacional de Vigilância Sanitária (Anvisa) ocorre em reunião pública ordinária da Diretoria Colegiada realizada nesta quarta-feira (17), que aprovou, por unanimidade, modificações da norma que trata das medidas a serem adotadas em aeroportos e aeronaves devido à Covid-19. A decisão manteve o desembarque das aeronaves de forma ordenada por fileiras, para reduzir aglomerações no corredor e o risco de contágio. A Anvisa também define a manutenção da oferta de álcool em gel em aeroportos e aeronaves, procedimentos de limpeza e desinfecção dos aviões, e sistemas de climatização. Os avisos sonoros serão mantidos, mas ajustados ao cenário pandêmico atual, incluindo a recomendação do uso de máscara por populações mais vulneráveis à Covid-19, como pessoas imunossuprimidas, crianças e idosos.
 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil A Agência Nacional de Vigilância Sanitária (Anvisa) recebeu o segundo pedido de registro de kit para teste para monkeypox, a varíola dos macacos. O pedido é para o produto Monkeypox Virus Nucleic Acid Detection Kit e foi apresentado pela empresa Comércio e Indústria de Produtos Médico-Hospitalares e Odontológicos Ltda (CPMH). De acordo com a agência reguladora, o pedido foi solicitado no dia 2 de agosto e já está em análise pela equipe técnica. Anteriormente, a Anvisa já havia o pedido de registro da empresa Biomédica. A solicitação foi analisada e a reguladora emitiu exigência, que é um pedido de informações e dados necessários para a conclusão da análise pela equipe técnica. O processo do registro envolve avaliar fabricação, confiabilidade dos resultados e efetividade para o diagnóstico.
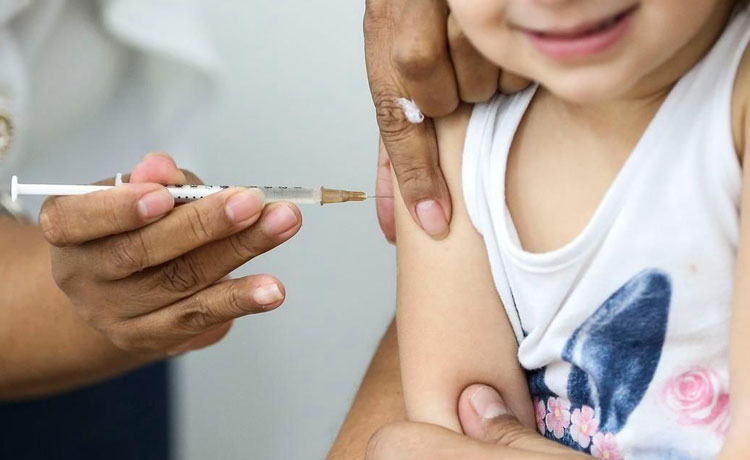 Foto: Marcelo Camargo/Agência Brasil
Foto: Marcelo Camargo/Agência Brasil A farmacêutica americana Pfizer submeteu, nesta sexta-feira (29), à Agência Nacional de Vigilância Sanitária (Anvisa), um pedido de aprovação para o uso da vacina contra a Covid-19 em crianças de 6 meses a 4 anos (4 anos, 11 meses e 29 dias). O imunizante foi aprovado para a faixa etária nos Estados Unidos em junho. No Brasil, a farmacêutica já imuniza pessoas acima dos 5 anos de idade. O pedido da Pfizer se apoia em um estudo que incluiu 4.526 crianças de 6 meses a 4 anos. Na pesquisa, as crianças receberam três doses (na quantidade de um terço a dose para o adulto), com um intervalo de três semanas entre a primeira e a segunda dose. A terceira aplicação foi administrada oito semanas após a segunda, em um momento em que Ômicron era a variante predominante. A formulação, segundo a Pfizer, é a mesma da vacina administrada nas demais faixas. A mudança seria na concentração, que é de 10 µg (micrograma) por dose na formulação pediátrica para crianças entre 5 a 11 anos, e de 3 µg por dose para crianças de 6 meses a 4 anos. Se aprovada, a vacina de 6 meses a 4 anos de idade terá uma tampa cor vinho para diferenciá-la da vacina pediátrica para crianças de 5 a 11 anos, que tem a tampa laranja, e do imunizante para a população acima dos 12 anos, cuja tampa é roxa.
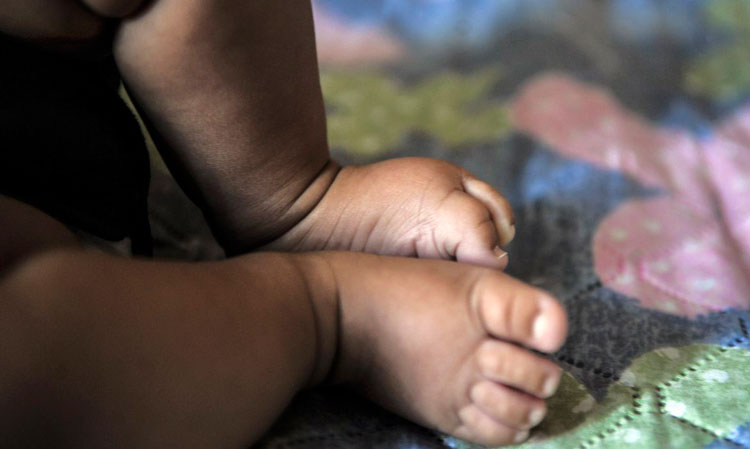 Foto: Marcello Casal Jr/Agência Brasil Saúde
Foto: Marcello Casal Jr/Agência Brasil Saúde Desde março de 2020, quando a covid-19 começou a se disseminar no Brasil, a doença causou no país a morte de 539 crianças entre 6 meses e 3 anos de idade. Esse número, atingido em pouco mais de dois anos de pandemia, é mais que o triplo do total de mortes que outras 14 enfermidades causaram juntas em um período de 10 anos. De acordo com a Agência Brasil, entre 2012 e 2021, neurotuberculose, tuberculose miliar, tétano neonatal, tétano, difteria, coqueluche, poliomielite, sarampo, rubéola, hepatite B, caxumba, rubéola congênita, hepatite viral congênita e meningite meningocócica do tipo B tiraram a vida de 144 crianças entre 6 meses e 3 anos de idade. Embora sejam enfermidades capazes de matar, todas elas podem ser prevenidas por vacinas. O levantamento, divulgado nesta segunda-feira (25), foi realizado por pesquisadores do Observatório de Saúde na Infância da Fundação Oswaldo Cruz (Fiocruz). Eles fizeram a comparação a partir de dados do Sistema de Informação sobre Mortalidade (SIM), desenvolvido pelo Ministério da Saúde. As 14 doenças consideradas no levantamento fazem parte da Lista Brasileira de Mortes Evitáveis para menores de 5 anos. Trata-se de uma relação criada por especialistas da saúde infantil sob a coordenação do Ministério da Saúde. Algumas dessas enfermidades não causaram nenhuma morte infantil nos últimos 10 anos. Um exemplo é a poliomielite, erradicada no país desde 1994. Diferente das 14 doenças, não há ainda um imunizante contra a covid-19 aprovado para a faixa etária estudada. Há duas semanas, a Agência Nacional de Vigilância Sanitária (Anvisa) autorizou o uso da vacina CoronaVac em crianças entre 3 e 5 anos de idade. Aquelas com mais de 5 anos já estavam sendo atendidas no Plano Nacional de Imunização (PNI) desde janeiro.





Achei Sudoeste © 2026 - Todos os direitos reservados.














